解析医疗器械备案分类 从第一类到第二类的监管演进与差异

医疗器械的备案与注册是保障其安全有效、规范市场准入的关键环节。在我国,根据风险等级,医疗器械被分为三类,其中第一类和第二类的备案与管理流程各有侧重,体现了“风险分级、科学监管”的原则。以“第一类医疗器械备案信息表-20210802072857”为例,该编号通常代表某具体产品在备案系统中的唯一标识,而“第二类医疗器械”则指向了更高风险等级的管理范畴。本文将系统梳理两者在备案要求、监管路径及市场意义等方面的核心差异。
第一类医疗器械备案:基于常规管理的低风险产品
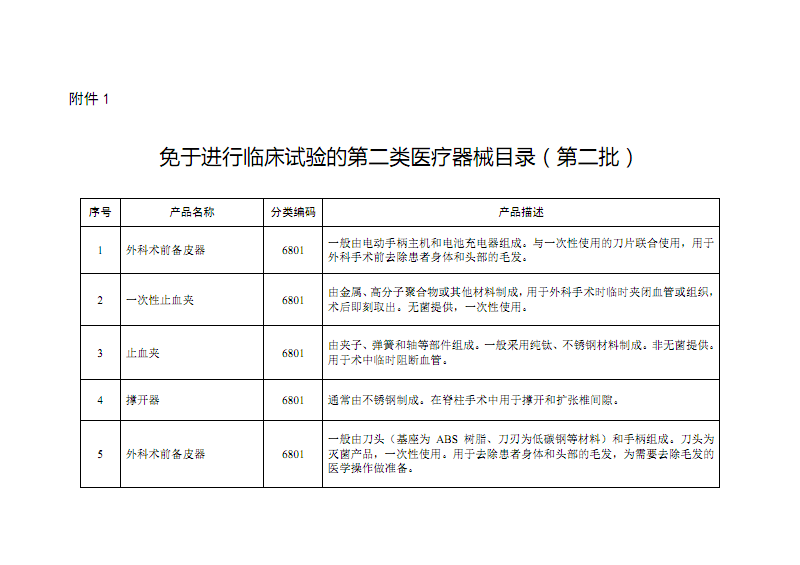
第一类医疗器械是指风险程度低,实行常规管理足以保证其安全有效的器械,如外科手术器械(非无菌)、听诊器、病床等。其备案流程相对简化,通常由备案人向所在地设区的市级药品监督管理部门提交备案资料,监管部门对资料进行形式审查,符合要求的予以备案,并发放备案凭证(即备案号,如示例中的“20210802072857”)。备案信息表一般包含产品名称、型号规格、备案人及生产企业信息、产品描述、预期用途等核心内容。备案制度的优势在于效率高、成本较低,有利于鼓励低风险产品的创新与快速上市。
第二类医疗器械注册:基于严格控制的中风险产品
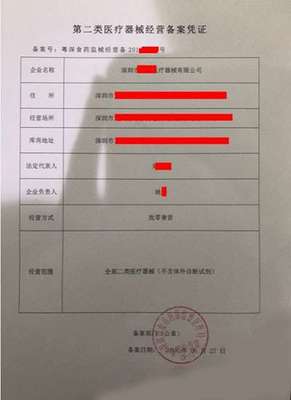
第二类医疗器械是指具有中度风险,需要严格控制管理以保证其安全有效的器械,如血压计、体温计、无菌手术刀、医用缝合针等。与第一类的“备案制”不同,第二类实行“注册制”。生产企业需向所在地省、自治区、直辖市药品监督管理部门提交产品技术资料、临床评价资料(部分产品可豁免)、风险管理资料等,经过技术审评、体系核查等严格程序,获得医疗器械注册证后方可生产销售。其监管强度、技术要求和审评周期都显著高于第一类备案。
核心差异与监管逻辑
- 管理模式:第一类为备案管理,侧重事后监督;第二类为注册管理,强调事前准入审批。
- 技术要求:第一类备案通常不要求提交详细的临床评价资料;第二类注册则大多需要提供临床评价或同品种比对资料,证明其安全有效性。
- 责任主体:第一类备案人/生产企业对备案资料的真实性、完整性负责;第二类注册申请人需承担更全面的产品全生命周期质量管理责任。
- 标识与追溯:两者均有唯一性标识(备案号/注册证号),但第二类产品的追溯体系要求通常更为严格。
市场意义与行业展望
“第一类医疗器械备案信息表-20210802072857”这样的具体备案实例,反映了我国医疗器械监管数据库的完善与透明度提升。而第二类医疗器械的严格注册流程,则是保障公众用械安全的重要防线。随着医疗器械产业创新加速和监管科学的发展,分类规则和备案注册要求也在动态优化。例如,部分低风险二类产品可能通过目录调整转入一类管理,以进一步激发市场活力;监管科技的应用也使得备案与注册信息的公开、共享与追溯更为高效。
从第一类备案到第二类注册,体现了医疗器械监管从低风险到中高风险的全链条、差异化治理思路。无论是备案凭证还是注册证号,都是产品合法上市的身份证明,共同构筑了公众健康的安全屏障。企业需精准把握产品分类,合规完成备案或注册,方能行稳致远。
如若转载,请注明出处:http://www.chinaskinhospital.com/product/47.html
更新时间:2026-06-19 16:15:44